Hirschsprungova choroba u dětí není tak častá, ale velmi závažná, a proto je zmatek rodičů, kteří slyší takovou diagnózu, zcela pochopitelný. Název nemoci většina maminek a otců ani neslyší, a proto existuje více otázek o této nemoci než odpovědí.
Co to je?
Hirschsprungova choroba dostala své jméno podle příjmení pediatra Haralda Hirschsprunga, který nejprve popsal tuto nemoc po smrti dvou svých mladých pacientů na těžkou chronickou zácpu. Tato nemoc má také jiná jména - aganglióza tlustého střeva nebo vrozený megakolon.
Je třeba poznamenat, že onemocnění je vrozenou anomálií ve vývoji tlustého střeva, při které je defekace extrémně obtížná kvůli narušení inervace (nervová spojení nebo nervová struktura) v jedné z částí tlustého střeva, častěji v nižších. V důsledku narušení nervové aktivity v tomto oddělení dochází k poklesu peristaltiky až do jeho úplné nepřítomnosti a v částech střeva nad postiženou zónou, ukládání velkého množství výkalů.
Podle WHO se Hirschsprungova choroba v dětství vyskytuje u jednoho z 5 000 novorozenců. Existují další údaje, které naznačují, že k onemocnění dochází s frekvencí 1: 2000 novorozenců. Chlapci jsou náchylnější k vrozeným chorobám - pro jednu nemocnou dívku jsou čtyři malí představitelé silnějšího pohlaví.
Podle statistik se u 9 z 10 dětí nachází vrozený megakolon před dosažením věku 10 let. Téměř v každém třetím případě je anomálie vývoje tlustého střeva doprovázena některými dalšími malformacemi a každé desáté dítě s takovou anomálií má Downov syndrom.


Proč se to stalo?
Vada je vždy vrozená, ale důvody, které ji způsobují, jsou stále předmětem studia lékařů a vědců. Dnes se odborníci domnívají, že vznik anomálie ovlivňuje několik faktorů najednou, z nichž hlavní je poškození několika genů najednou, které jsou ve skutečnosti zodpovědné za to, jak správně a včas budou vytvořeny nervové struktury stěn tlustého střeva. V genetice je vše velmi komplikované a pro rodiče může být obtížné pochopit, proč měli zdravé dítě nemocné. Zkusme to ale vysvětlit.
Dědičný faktor. Přibližně každé páté dítě s vrozeným megakolonem má rodinný genetický vliv. Během koncepce se vytvoří proces mutace v genech RET, GDNF, EDN3, ENDRB. V 7-12 týdnech těhotenství, kdy plod dokončí tvorbu nervových buněk ve stěnách vnitřních orgánů, nemohou uvedené postižené geny zajistit přechod zárodečných buněk - neuroblastů (ze kterých se tvoří neurony) na požadované místo. Asi v 11–12% případů genetika nalezla v chromozomech aberace (nevysvětlitelné chyby). A u každého pátého dítěte s Hirschsprungovou chorobou je anomálie úzce spojena s jakýmsi dědičným syndromem.
Anomálie na úrovni nitroděložního vývoje. Vznikají u dětí, které v době početí měly zdravé geny uvedené výše, ale problém a omyl vznikly v procesu embryogeneze. Důvodem může být virová infekce, chřipka, kterou matka utrpěla v rané fázi těhotenství, účinek vysokých dávek radioaktivního záření na tělo těhotné ženy během tohoto období, kontakt ženy v raných stádiích s látkami, které mohou způsobit genetické mutace plodu. V tomto případě byla distribuce neuroblastů zárodečných buněk narušena.
Lékaři sledovali statistiky a s jistotou říkají, že pravděpodobnost Hirschsprungovy choroby u dětí se zvyšuje, pokud těhotná žena trpí různými porodnickými patologiemi, gynekologickými chorobami, stejně jako gestózou, diabetes mellitus, hypertenzí, srdečním selháním nebo některými srdečními chorobami.


Je to období od 7 do 12 týdnů těhotenství, které je považováno za kritické z hlediska pravděpodobnosti selhání tvorby nervových struktur stěn tlustého střeva plodu. Během tohoto období byly položeny takzvané Meissnerovy nervové plexy, které se nacházejí u zdravých lidí ve vrstvě tlustého střeva pod sliznicí. Ve stejném období dochází k tvorbě uzlů a plexusů nervů nazývaných Auenbachův plexus, který se nachází ve svalové vrstvě střeva.
Vzhledem k tomu, že embryonální neuroblasty nedorazí na správné místo včas nebo nepřijdou v dostatečném množství, jsou tam vytvořena nervová zakončení s chybami - buď jejich jednotlivé fragmenty, nebo nevyvinutá slabá a nesprávná síť. Tato oblast bez normálního nervového zásobení se nedokáže vyrovnat s hlavním úkolem tlustého střeva - odstraněním obsahu ven. Proto dochází k neustálé zácpě a pak se začnou rozšiřovat horní části střeva.


Klasifikace a typy patologie
Lékaři budou hovořit o typu a typu onemocnění u novorozence, dítěte nebo staršího dítěte na základě anatomických a klinických rysů anomálie u konkrétního dítěte.
Hlavním kritériem je délka postižené oblasti střeva.
Nejběžnějším případem je rektosigmoidální forma. Vyskytuje se u 65-75% pacientů. V každém čtvrtém případě dochází k rektální formě onemocnění. Pouze ve 3% případů je nalezena mezisoučetní forma, v 1,5% případů má léze segmentální povahu. Celková forma, ve které je postiženo celé tlusté střevo a je zbaveno nervové podpory, se vyskytuje v 0,5% případů.
Jak již bylo zmíněno, horní části se rozšiřují kvůli neustálému hromadění fekálních hmot a v závislosti na přesném místě patologické expanze se ke specifické diagnóze přidávají označení místa expanze jako megarectum, megasigma, subototální a celkový megakolon atd.


Kombinace příznaků a projevů vývojových abnormalit umožňuje lékařům identifikovat několik typů onemocnění.
- Typické dítě - vyskytuje se v 90-95% případů. Vyvíjí se rychle, je doprovázen velmi častou zácpou, kdy bez vnější pomoci dítě prakticky nedokáže vyprázdnit střeva, rychle se objeví první příznaky střevní obstrukce.
- Přetrvávající dítě - poprvé se objeví po kojeneckém věku. Obvykle je to způsobeno skutečností, že je ovlivněna malá oblast střeva, a proto se klinický obraz vyvíjí pomalu, prodlouženě.
- Skrytý - poprvé se bolestivá zácpa stává problémem blíže adolescentnímu období vývoje dítěte. Střevní obstrukce se rychle zvyšuje.
Rozlišují se také kompenzované, subkompenzované a dekompenzované formy onemocnění. Pokud se tělu podaří problém částečně kompenzovat, může se dítě v průběhu let vyprázdnit, ale postupně bude náchylné k zácpě, jejíž délka se může pohybovat od 3 dnů do týdne. U subkompenzované Hirschsprungovy choroby je dítě schopné vyprázdnění, ale pouze za použití klystýru nebo projímadla. Bez nich, zácpa.
Ve stavu dekompenzace se dítě nenakopává a ani nepociťuje nutkání k tomuto přirozenému očistnému procesu. Ztluštění stolice ve střevech se stává kamenným a laxativa a klystýry nepomáhají.


Příznaky a příznaky
Závisí to na tom, jak moc je postiženo tlusté střevo, kdy bude možné určit první příznaky patologie u dítěte. V naprosté většině případů je anomálie stanovena po narození, ale není vyloučen latentní průběh s menšími příznaky, což umožní diagnostikovat Hirschsprungovu chorobu pouze ve škole, dospívání nebo později.
Projevuje se inervace tlustého střeva chronická zácpa. Pokud má dítě nutkání vyprázdnit střeva, ale nedokáže to, nemluvíme o vrozené anomálii. S vrozeným megakolonem dítě obvykle nepociťuje nutkání vyprázdnit střeva. Téměř polovina dětí s touto diagnózou trpí nadýmáním a bolestmi břicha spojenými s hromaděním výkalů a expanzí horních střev.
Dlouhá přítomnost anomálie tvoří zjevnou asymetrii břicha. Modré žíly jsou viditelné na břiše pouhým okem. Dlouhý průběh onemocnění vede u dítěte k anémii, jeho pokožka je bledá, trpí častými závratěmi a rychle se unaví. Kvůli hromadění výkalů mohou existovat příznaky intoxikace, které se projevují bolestmi hlavy, nevolností a někdy zvracením.


Různé fáze mají různé vlastnosti.
- Kompenzováno - zácpa u novorozenců, která se zhoršuje zavedením doplňkových potravin. Pokud rodiče sledují frekvenci stolice, dávají klystýry včas, pak není narušen celkový stav dítěte. Ve druhém stupni začíná trpět dětská chuť k jídlu.
- Subkompenzováno - zácpa se stává častější, bez klystýru je téměř nemožné vyprázdnit střeva, ale při adekvátní léčbě má patologie každou šanci jít do kompenzované formy. Příznaky Hirschsprungovy choroby rostou pomalu a postupně.
- Dekompenzováno - Příznaky se hromadí velmi rychle. Na prvním stupni je patrný akutní průběh - dítě má zhoršené vypouštění plynů, nedochází k pohybu střev, břicho je oteklé jako „žába“. Ve druhém stupni se onemocnění stává chronickým.
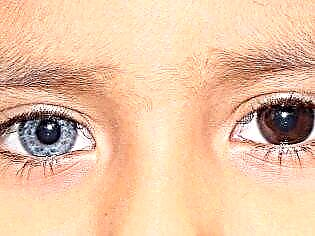
Zajímavý detail: vrozený megakolon se často vyskytuje u dětí, které mají heterochromii od narození - jinou barvu duhovky orgánů vidění.

Jaké je nebezpečí?
Aganglióza tlustého střeva je nebezpečná, protože jsou narušeny zažívací procesy nemocného dítěte, živiny jsou absorbovány méně, než je nutné, a protože k tomu dochází na pozadí snížení chuti k jídlu, je možné, že se dítě stane hypotrofickým nebo začne trpět extrémním vyčerpáním - kachexií.
Děti s takovou vrozenou vadou rostou pomalu, zaostávají za svými vrstevníky ve výšce a hmotnosti a často trpí dysbiózou, zánětem střevních sliznic.
Nejnebezpečnější komplikací je vývoj střevní obstrukce, stejně jako tvorba dekubitů střevních stěn s fekálními kameny v rozporu s celistvostí samotného střeva. Je nebezpečný při peritonitidě a sepse.


Diagnostika
Lékař může navrhnout, že dítě má střevní agangliózu na základě stížností rodičů na časté nadýmání, potíže s defekací a zácpu. Když se zdravotník dotkne prstů břicha, provedou se dva testy - test „nádoru“ a test „jílovitých příznaků“. V prvním případě lékař objeví těsnění při digitálním vyšetření břicha a ve druhém příznaky stlačení tlustého střeva. V tomto případě je dítě odesláno k vyšetření, které je určeno k potvrzení nebo popření diagnózy. To zahrnuje:
- RTG tlustého střeva naplněný kontrastní látkou;
- endoskopie konečníku a sigmoidního tračníku;
- cytomorfologie podle Swensona - histologické vyšetření vzorků tkáně konečníku a tlustého střeva;
- rozchodové zkoušky.
Při analýze krve dítěte s Hirschsprungovou chorobou se obvykle nacházejí zvýšené leukocyty, ESR a toxické změny neutrofilů.
Provádí se vyšetření a diagnostika proktology, chirurgové, specialisté na infekční nemoci, pediatři.

Léčba a prognóza
U dítěte s touto diagnózou se obvykle doporučuje chirurgický zákrok. A rodiče by měli pochopit, že pokud to lékaři doporučí, je lepší to neodmítnout, protože Hirschsprungova nemoc není léčena konzervativně a lidovými metodami. Během operace bude možné obnovit průchodnost střeva a odstranit postiženou oblast, což vytváří tolik potíží v práci celého střeva.
Existují dva typy operací - jednostupňový a dvoustupňový. Abychom pochopili, který z nich doporučit, lékař bere v úvahu věk dítěte, stupeň poškození střev, délku inervované oblasti.
Jednostupňová operace se obvykle provádí s kompenzovaným onemocněním a drobným poškozením střev. Jednoduše odstraňte postiženou oblast a vytvořte anastomózu. Tato metoda je méně traumatizující, soudě podle recenzí lékařů a pacientů.
Dvoustupňová operace se provádí u dětí s onemocněním ve stadiu subkompenzace a dekompenzace s velkými postiženými oblastmi tlustého střeva. První fáze zahrnuje odstranění postižené oblasti a vytvoření dočasné kolostomie. Ve druhé fázi se z ní vytvoří anastomóza.

Nouzovou operaci dítěte lze provést v případě střevní obstrukce, proleženin stěn, porušení integrity střevních stěn.
Je docela těžké něco předvídat koneckonců vše závisí na tom, jak brzy bylo dítěti diagnostikována vrozená anomálie vývoje, k jakému typu patří, zda byla všechna klinická doporučení dodržena včas. Při včasném provozu jsou prognózy příznivé. Pokud se kojenec neléčí, odhaduje se pravděpodobnost úmrtí dítěte na 80%.
Je-li od operace upuštěno a vytvoří se následující překážka, je možné, že po operaci dostane dítě zdravotní postižení, ale i zde je vše individuální a záleží na objemu chirurgického zákroku.

Přečtěte si více o této nemoci v dalším videu.